| Manual Pipeline V3
1- Data upload
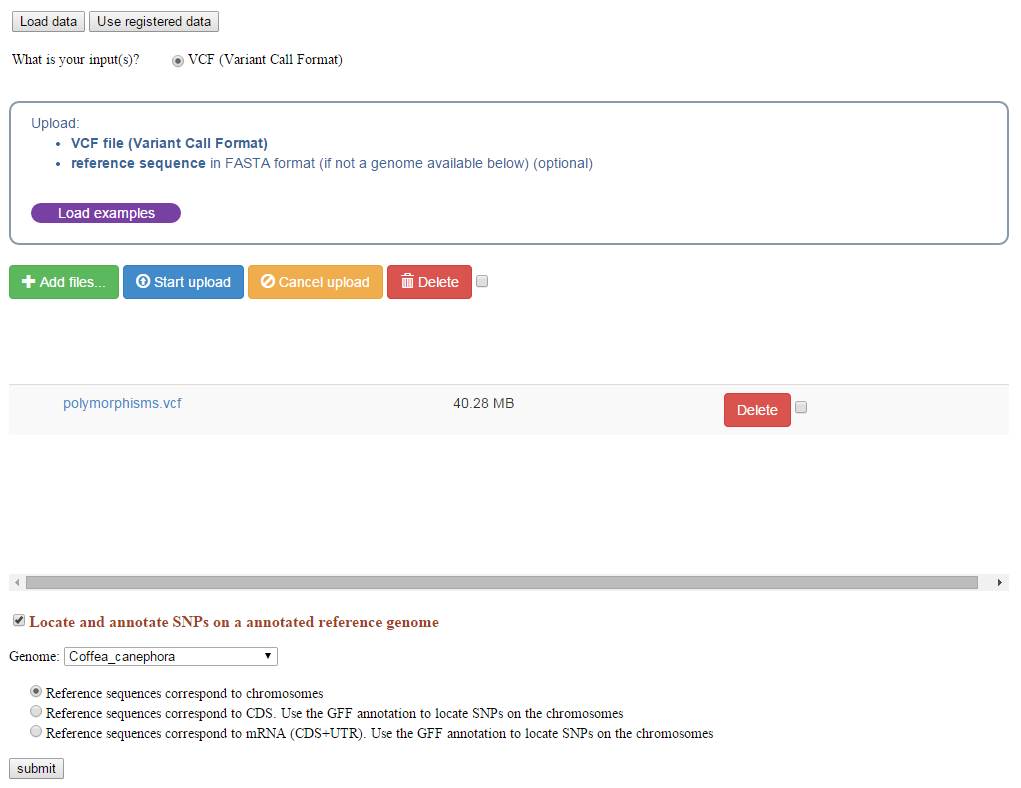
The process takes VCF genotyping files as input. VCF stands for Variant Call Format, this file format was initially used by the 1000 Genomes project to encode SNPs and other structural genetic variants. This format is further described on the 1000 Genomes project Web site, and be can obtained by SNP calling softwares such as GATK.
Users may then select the reference genome on which the SNP calling was performed. 14 reference genomes are available.
The reference sequence can also be provided and uploaded as a Fasta sequence file.
Using the reference genome and the associated GFF annotation file, the process computes the Snpeff program to annotate the SNPs (intron/exon location and effect prediction).
By default, the process assumes that the VCF file is based on chromosomal position. If not, an option allows to recalculate the chromosomal positions (before launching Snpeff) from positions of variants established within CDS or complete mRNA (for instance when NGS reads were mapped against the predicted transcriptome).
2- Variant filtering
The process extracts the different individuals and reference sequences from the VCF file and offers an intuitive interface for SNP filtering.
Variants can be filtered and selected using various criteria (Minor Allele Frequency, SNP annotation, missing data...).
All filters are applied on the selected subsets of individuals (on the left) and of reference sequences (on the right).
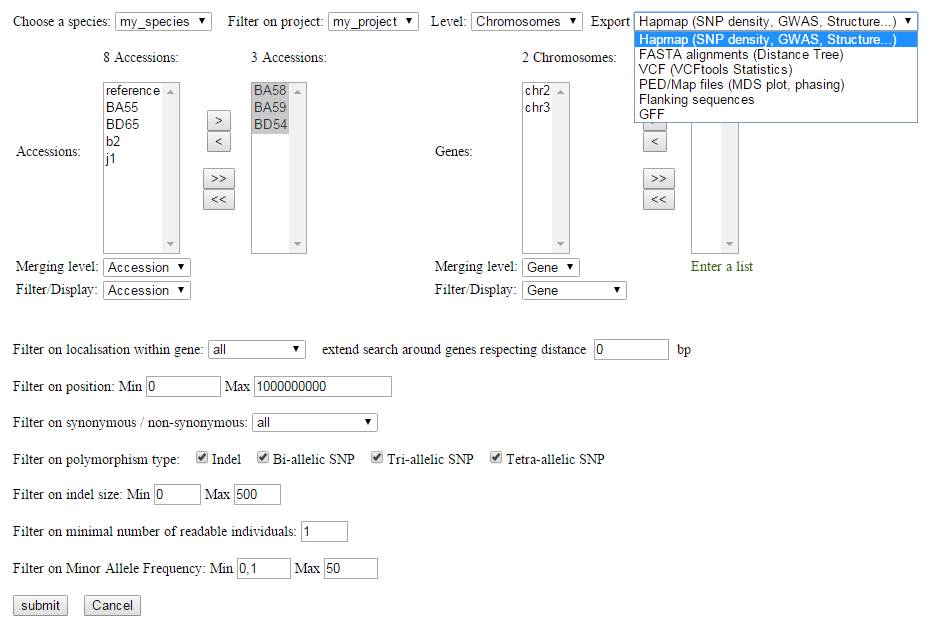
According to the selected output format, the process allows the variants and the genotyping matrix to be sent to a specific type of analysis:
- Hapmap format for GWAS (Genome Wide Association Studies), SNP density or Population structure
- VCF format for general statistics or diversity indexes along the genome
- Fasta format for distance tree
- PED and Map (Plink compatible) format for MDS plot (Multi-Dimensional Scaling)
- GFF format
- Flanking sequences to be sent for chip design
|
|

